Медицински стручњак за чланак

Нове публикације
Синдром Пјера Робина
Последње прегледано: 04.07.2025

Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.

Пјер-Робинов синдром, познат у медицини и као Робинова аномалија, је конгенитална патологија развоја виличног дела лица. Болест је добила име у част француског стоматолога П. Робина, који је први описао све њене знаке. Ланелонг и Менар су први описали Пјер-Робинов синдром 1891. године у свом извештају о два пацијента са микрогнатијом, расцепом непца и ретроглосоптозом. Године 1926, Пјер-Робин је објавио случај болести код одојчета са знацима класичног синдрома. До 1974. године, тријада знакова била је позната као Робин-Пјеров синдром. Међутим, овај синдром се сада користи за описивање малформација са истовременим присуством вишеструких аномалија.
Епидемиологија
То је хетерогена конгенитална мана која има преваленцију од 1 на 8.500 живорођених. Однос мушкараца и жена је 1:1, осим X-везаног облика.
Међу овим пацијентима, 50% новорођенчади има непотпуну расцеп меког непца, остатак се рађа са лучним и необично високим непцем, али без расцепа.
Узроци Синдром Пјера Робина
Разматра се могућност аутозомно рецесивног наслеђивања болести. Постоје две врсте синдрома у зависности од етиологије: изоловани и генетски детерминисани. Изоловани тип се развија услед компресије доњег дела вилице током ембрионалног развоја. Компресија се може развити због:
- Присуство локализованих печата у материци (цисте, ожиљци, тумори).
- Вишеструка трудноћа.
Такође, развој вилице код фетуса може бити поремећен:
- Вирусне инфекције које је будућа мајка претрпела током трудноће.
- Неуротрофични поремећаји.
- Недовољна количина фолне киселине у телу труднице.
Патогенеза
Пјер Робинов синдром је узрокован ембрионалним поремећајима који су узроковани широким спектром патологија у пренаталном периоду.
Постоје три патофизиолошке теорије које могу објаснити појаву Пјер Робиновог синдрома.
Механичка теорија: Ова теорија је највероватнија. Неразвијеност мандибуларног апарата јавља се између 7. и 11. недеље трудноће. Висок положај језика у усној дупљи доводи до стварања пукотина на непцу, због којих се шупља вена не затвара. Ова теорија објашњава класичну пукотину у облику обрнутог слова U и одсуство придружене расцепљене усне. Олигохидрамнион може играти улогу у етиологији, јер одсуство амнионске течности може довести до деформације браде и накнадне компресије језика између шупљих вена.
Неуролошка теорија: Кашњење у неуролошком развоју је примећено електромиографијом мишића увуле и фарингеалних стубова, као и осетљивошћу укуса због кашњења проводљивости у хипоглосалном нерву.
Теорија диснеурорегулације ромбенцефалона: Ова теорија се заснива на поремећају развоја ромбенцефалона током онтогенезе.
Недовољан развој доњег дела вилице детета доводи до тога да се усна дупља значајно смањује. То, заузврат, узрокује такозвану псеудомакроглосију, односно језик је померен на задњи део фарингеалног зида. Ова патологија доводи до развоја опструкције дисајних путева.
Докле год беба плаче или се креће, дисајни путеви остају проходни, али чим беба заспи, поново долази до опструкције.
Због респираторних поремећаја, процес храњења бебе је веома тежак. У овом тренутку готово увек долази до опструкције дисајних путева. Ако се не примени медицинска корекција, таква патологија може довести до тешке исцрпљености целог тела, па чак и смрти.
Симптоми Синдром Пјера Робина
Болест карактеришу три главна симптома:
- Доња микрогнатија (неразвијеност доње вилице, јавља се у 91,7% случајева болести). Карактерише је повлачење доњег зубног лука за 10-12 мм иза горњег лука. Доња вилица има мало тело, туп угао. Дете постиже нормалан развој приближно са 5-6 година.
- Глосоптоза (повлачење језика због његовог недовољног развоја, примећено у 70-85% случајева).
- Макроглосија и анкилоглосија су релативно ретки симптоми, примећени у 10-15% случајева.
- На небу се појављује пукотина.
- Брадипнеја и диспнеја.
- Блага цијаноза.
- Асфиксија, која се најчешће јавља током покушаја храњења бебе.
- Гутање је немогуће или веома тешко.
- Осећај као да повраћа.
- Аурикуларне аномалије у 75% случајева.
- Кондуктивни губитак слуха јавља се код 60% пацијената, док се атрезија спољашњег слушног канала јавља само код 5% пацијената, недовољна пнеуматизација мастоидне шупљине темпоралне кости.
- Аномалије унутрашњег уха (аплазија латералних полукружних канала, велики вестибуларни аквадукт, губитак кохлеарних длакавих ћелија).
- Назалне малформације су ретке и састоје се углавном од аномалија корена носа.
- Зубне малформације се јављају у 30% случајева. Ларингомалација и велофарингеална инсуфицијенција се јављају код приближно 10-15% пацијената са Пјер Робеновим синдромом.
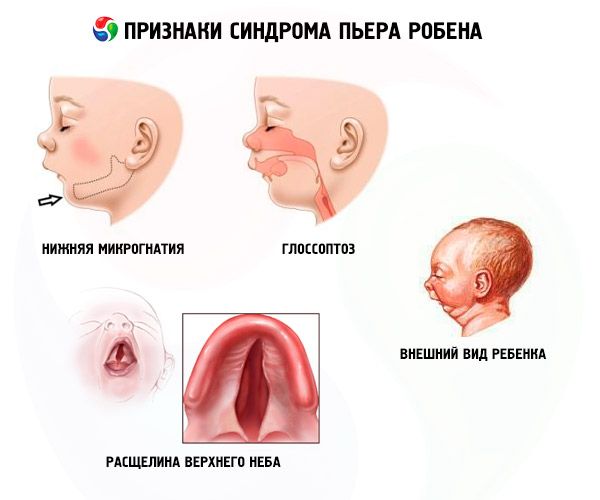
Системске карактеристике Пјер Робин синдрома
Системске развојне аномалије су описане у 10-85% регистрованих случајева.
Абнормалности ока јављају се код 10-30% пацијената. Оне могу укључивати: хиперопију, миопију, астигматизам, склерозу рожњаче и стенозу назолакрималног канала.
Кардиоваскуларне патологије: бенигни шумови на срцу, стеноза плућне артерије, отворени дуктус артериозус, овални прозор, дефект атријалног септума и плућна хипертензија. Њихова преваленција варира од 5-58%.
Аномалије повезане са мишићно-скелетним системом (70-80% случајева): синдактилија, диспластичне фаланге, полидактилија, клинодактилија, хипермобилност зглобова и олигодактилија горњих удова. Аномалије доњих удова: аномалије стопала (клинасто стопало, метатарзална аддукција), фемурне малформације (валгусна или варусна карлица, кратки фемури), аномалије кука (конгенитална дислокација, контрактуре), аномалије коленског зглоба (ГЕНУ ВАЛГУС, синхондроза). Малформације кичменог стуба: сколиоза, кифоза, лордоза, вертебрална дисплазија, агенеза сакрума и кокигеалног синуса.
Патологија централног нервног система: епилепсија, кашњења у развоју нервног система, хидроцефалус. Учесталост дефеката ЦНС-а је око 50%.
Генитоуринарне аномалије: неспуштени тестиси (25%), хидронефроза (15%) и хидроцела (10%).
Повезани синдроми и стања: Стиклеров синдром, синдром трисомије 11q, трисомија 18, синдром делеције 4q, реуматоидна артропатија, хипохондроплазија, Мебијусов синдром.
Фазе
Постоје три фазе тежине болести, које зависе од стања респираторног тракта детета:
- Благо - постоје мањи проблеми са храњењем, али дисање готово да није отежано. Лечење се спроводи амбулантно.
- Умерено – дисање је умерено отежано, храњење детета је умерено отежано. Лечење се спроводи у болници.
- Тешко – дисање је веома отежано, дете не може нормално да се храни. Потребна је употреба посебних уређаја (интраназална сонда).
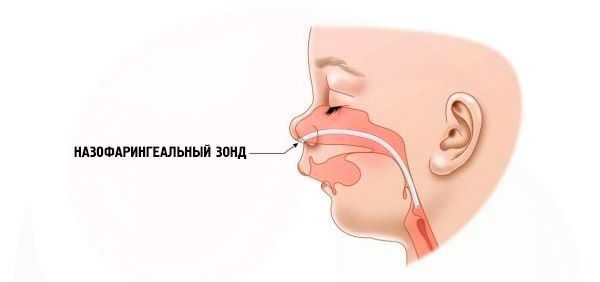
Компликације и посљедице
Комбинација микрогнатије и глосоптозе може довести до тешких респираторних компликација и проблема током храњења детета.
Пјер Робинов синдром изазива следеће компликације:
- Стридозно дисање због опструкције дисајних путева. Ларингомалација или чак асфиксија у сну.
- Психомоторни развој детета знатно заостаје за његовим вршњацима.
- Физички развој такође заостаје.
- Говор пацијената је оштећен.
- Честе инфекције уха које постају хроничне и доводе до оштећења слуха.
- Синдром опструктивне апнеје у сну, појава смрти у сну варира у 14-91% случајева.
- Проблеми са зубима.
Дијагностика Синдром Пјера Робина
Дијагноза Пјер Робиновог синдрома није тешка. Заснива се на клиничким манифестацијама. Да би се искључиле друге патологије, веома је важно консултовати генетичара.
Деца са Робиновом конгениталном аномалијом имају проблема са дисањем од рођења због сталног повлачења језика уназад. Беба је немирна, кожа јој је плавкаста, звиждање излази из грудног коша приликом удисања. Током храњења може доћи до гушења. Дијагноза се може поставити и по необичном изгледу детета - „птичје лице“. Често се код пацијената развијају и други дефекти: миопија, катаракта, патологија генитоуринарног система, патологија срца, аномалије у развоју кичме.
На основу ових клиничких манифестација, специјалисти неће бити тешко да постави тачну дијагнозу.
Кога треба контактирати?
Третман Синдром Пјера Робина
Лечење се спроводи одмах након рођења детета са Пјер Робиновим синдромом. Ако је болест блага, онда је за побољшање стања пацијента потребно стално држати дете вертикално или лежећи на стомаку. Глава бебе треба да буде нагнута ка грудима. Током храњења се не препоручује држање детета у хоризонталном положају како храна не би доспела у респираторни тракт.
Ако је неразвијеност доње вилице прилично изражена, користи се хируршка интервенција да би се увучени језик вратио у нормалан физиолошки положај. У тешким случајевима, језик се повлачи нагоре и фиксира на доњој усној. У веома тешким случајевима, мора се извршити трахеостомија, глосопексија и дистракциона остеогенеза доње вилице.
Такође се користи конзервативни третман.
Лекови
Фенобарбитал. Лек за спавање и седатив, има антиконвулзивно дејство. Свака таблета садржи 100 мл фенобарбитала. Дозирање је индивидуално, јер зависи од тежине болести и стања детета. Лек је забрањен за пацијенте са отказивањем јетре, хиперкинезом, анемијом, мијастенијом, порфиријом, дијабетес мелитусом, депресијом и нетолеранцијом на компоненте. Приликом узимања могући су следећи симптоми: вртоглавица, астенија, халуцинације, агранулоцитоза, мучнина, низак крвни притисак и алергије.
Клоназепам. Лек који се прописује за лечење епилепсије. Лек садржи активну супстанцу клоназепам, који је дериват бензодиазепина. Има антиконвулзивно, анксиолитичко и мишићно релаксирајуће дејство. Дозу одређује лекар који лечи лек, али не сме прећи максимум - 250 мцг дневно. Не узимати у случају несанице, мишићне хипертоније, психомоторне агитације, паничних поремећаја. Приликом узимања могући су следећи симптоми: летаргија, мучнина, дисменореја, главобоља, леукопенија, задржавање или инконтиненција урина, алопеција, алергија.
Сибазон. Доступан у облику раствора и ректалних таблета. Активна супстанца је дериват бензодиазепина (сибазон). Има седативно, анксиолитичко, антиконвулзивно дејство. Дозирање је индивидуално. Пацијентима са хроничном хиперкапнијом, мијастенијом, интолеранцијом на бензодиазепине је забрањено узимање лека. Приликом употребе лека могу се развити следећи симптоми: мучнина, затвор, главобоља, вртоглавица, штуцање, уринарна инконтиненција, алергије.
Кортексин лиофилизат. Лек са ноотропним дејством. Лек садржи комплекс водорастворљивих полипептидних фракција и глицин. Дозирање је индивидуално и прописује га лекар у складу са стањем пацијента. Пацијентима са нетолеранцијом на кортексин је забрањено узимање лека. Лек може изазвати алергијске реакције.
Физиотерапијски третман
Типично, у благим фазама синдрома, користи се позициона терапија, где се дете поставља на стомак у усправном положају док гравитација не примора доњу вилицу да правилно расте.
Хируршко лечење
Хируршко лечење се првенствено користи за корекцију глосоптозе. Постоји неколико метода:
- Подупирање језика сребрним концем. Конац се провлачи кроз доњи део десни и доњу усну. Метода се назива Дагласова.
- Духамелова метода - дебела сребрна нит се провлачи кроз корен пацијентовог језика и оба образа. Користити не дуже од тридесет дана.
- Ортопедски уређаји за екстензију и фиксацију језика.
- Са годину дана старости може се извршити операција за исправљање расцепа непца.
Прогноза
Прогноза и ток болести су тешки. Најчешће, смрт наступа у првим данима живота у умереном и тешком стадијуму болести (узрок је асфиксија). Такође, ризик од смрти у првој години је прилично висок због бројних инфекција.
За пацијенте старије од две године, прогноза је повољна.
 [ 36 ]
[ 36 ]