
Усхеров синдром
Последње прегледано: 23.04.2024

Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.
Усхеров синдром је наследна болест која се манифестује у облику потпуне глухосте од рођења, као и прогресивног слепила са годинама. Губитак вида повезан је са ретинитисом пигмента - ово је процес пигментне дегенерације ретине очију. Многи људи са Усхеровим синдромом такође имају озбиљне проблеме са балансом.
Епидемиологија
Због истраживања, било је лако утврдити да је око 8% испитаних глувонемских деце болесно с Усхеровим синдромом (тестирање је спроведено у специјалним установама за глувонеме). Пигментирани ретинитис је примећен код 6-10% пацијената са урођеном глухостом, што је запажено код око 30% људи са пигментираном мрежњачком болести.
Сматра се да се ова болест манифестује у око 3-10 људи од 100 хиљада широм света. То се може посматрати и код жена и мушкараца. Овај синдром погађа око 5-6% светске популације. Око 10% свих случајева педијатријске дубоке глувоће се јавља услед синдрома Усхер И, а такође и типа ИИ.
У Сједињеним Државама типови 1 и 2 су најчешћи типови. Заједно, они чине око 90 до 95 процената свих случајева Усхеровог синдрома код деце.
Узроци усхеров синдром
Усхеров синдром И, ИИ, а такође и ИИИ врсте носи аутозомни рецесивни узрок, али се тип ИВ сматра кршењем Кс хромозома. Узроци овог синдрома слепила, као и глувоће, нису довољно проучавани. Претпоставља се да су људи са овом болестом преосетљиви на компоненте које могу оштетити структуру ДНК. Уз ову болест, поремећаји имунолошког система такође могу бити повезани, али у овом случају не постоји прецизна слика таквог процеса.
Године 1989. Пацијенти са болести типа ИИ први су дијагностиковали хромозомске абнормалности, што би могло довести до начина изолације гена који изазивају развој синдрома. Поред тога, такође ће бити могуће идентифицирати ове гене од својих носиоца и развити посебне антенаталне генетске тестове.
 [8]
[8]
Фактори ризика
Насљеђивање синдрома се јавља у случају када су оба родитеља болесна, односно наследство је рецесиван тип. Дете такође могу наследити болест у случају да су његови родитељи носиоци гена. Ако су оба родитеља има ген, онда је вероватноћа рођења њиховог детета са овим синдромом је 1 до 4. Особа која има само један синдром ген се сматра да је носилац, али он нема симптоме поремећаја. Данас још није могуће утврдити да ли особа има ген за ову болест.
Ако се дете роди родитељима, од којих један нема такав ген, вероватноћа да ће наследити овај синдром је врло низак, али ће он бити недвосмислен носилац.
Симптоми усхеров синдром
Симптоми Усхеровог синдрома су губитак слуха, а поред ове патолошке акумулације пигментисаних ћелија у структурама око. Даље, пацијент развија дегенерацију очне ретине, због чега почиње погоршање вида уз накнадни губитак у најтежим случајевима.
Сензоринеурални губитак слуха је блага или потпуна и обично не напредује од рођења. Али пигментоза ретинитиса може почети да се развија у детињству или касније. Резултати истраживања показали су да се острина централног вида може наставити дуги низ година, чак и када се периферни вид погоршава (овај услов се зове "тунелски вид").
Ово су главне манифестације болести, које понекад могу допунити и други поремећаји - као што су психоза и други ментални поремећаји, проблеми са унутрашњим ушима и / или катаракте.
Обрасци
Током истраживања идентификоване су 3 врсте ове болести, а такође и 4 форме - прилично ретко.
И врста болести карактерише урођена потпуна глувоћа, као и поремећај равнотеже. Често те дјеце почињу ходати само у доби од 1,5 године. Погоршање вида обично почиње са 10 година, а коначни развој стања ноћног слепила почиње са 20 година. Код деце са овом врстом болести може се развити прогресивно погоршање периферног вида.
Код болести типа ИИ примећена је умерена или урођена глувоћа. Често се у овом случају више не јавља погоршање делимичне глувоће. Ретинитис пигмента почиње да се развија око краја адолесцентног периода или после 20 година. Развој ноћног слепила обично почиње у 29-31 години. Поремећаји оштрине вида у случају патологије типа ИИ у основи напредују нешто спорије него у типу И.
Тип ИИИ болести које карактерише прогресивним губитком слуха, обично почиње у пубертету, као и постепено појављивање у истом периоду (мало касније него што глувоћа) ретинитис пигментоса да може да постане фактор у развоју прогресивне слепила.
Манифестације четвртог типа патологије углавном се јављају код мушкараца. У овом случају постоје и прогресивни поремећаји и губитак слуха и вида. Овај облик је веома реткост и обично има природу Кс-хромозома.
Дијагностика усхеров синдром
Дијагноза Усхеровог синдрома врши се на основу пацијентовог комбиновања изненадне глувоће са прогресивним губитком вида.
Анализе
Да би се открила мутација, може се одредити посебан генетски тест.
Пронађено је 11 генетских локуса које могу узроковати развој Усхеровог синдрома и идентификовале девет гена који су управо узрочник поремећаја:
- Тип 1: МИ07А, УСХ1Ц, Цдх23, Пцдх15, САНС.
- Тип 2: усх2а, ВЛГР1, ВХРН.
- Тип 3 Усхира синдром: УСХ3А.
НИДЦД научници, заједно са колегама са универзитета у Њујорку и Израела су идентификовали мутацију названу Р245Кс Пцдх15 ген, који је велики проценат типа 1 Усхер синдром код јеврејског становништва.
Да бисте сазнали о лабораторијама које спроводе клиничка испитивања, посетите хттпс://ввв.генетестс.орг и претражите каталог лабораторијских истраживања тако што ћете укуцати термин "Усхеров синдром".
Да бисте сазнали о актуелним клиничким студијама које укључују генетска тестирања Усхер синдром, посетите сајт и унесите претрагу хттпс://ввв.цлиницалтриалс.гов "Усхер синдром" или "Усхер синдром генетско тестирање."
[25], [26], [27], [28], [29], [30]
Инструментална дијагностика
Постоји неколико метода инструменталне дијагностике:
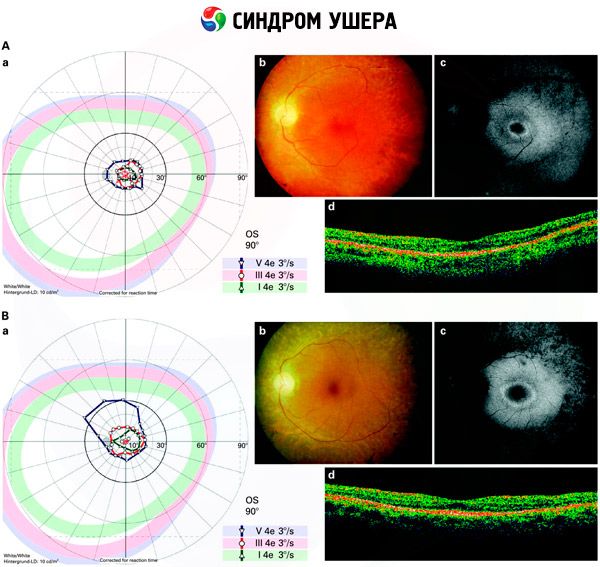
- Испитивање фундуса за идентификацију присутности ретина на мрежњачи, као и сужење мрежњачких ћелија;
- Електроретинограм, који вам омогућава да идентификујете почетне дегенеративне абнормалности у ретини очију. Приказује изумирање електро-радиографских стаза;
- Електронистагмограм (ЕНГ) мери нехотичне покрете очију, што би могло указати на присуство дисбаланса
- Аудиометрија, која одређује присуство глувоће и степен његове тежине.
Диференцијална дијагноза
Усхеров синдром се мора разликовати са неким сличним абнормалностима.
Синдром Халлгрена, у коме се примећује конгенитални губитак слуха, као и прогресивни губитак вида (појављују се и катаракте и нистагмус). Међу додатним симптомима болести: атаксија, психомоторни поремећаји, психоза и ментална ретардација.
Алстромов синдром, који је наследна болест у којој се јавља дегенерација ретине, због чега се губи централни вид. Овај синдром је повезан са проблемом гојазности детета. У овом случају, дијабетес мелитус и губитак слуха почињу да се развијају након 10 година.
Рубела код труднице у првом тромесечју може изазвати разне абнормалности у развоју дјетета. Међу последицама ове аномалије је губитак слуха, као и (или) проблеми са видом, а поред тога и разни развојни недостаци.
Кога треба контактирати?
Третман усхеров синдром
За излечење Усхеровог синдрома је сада немогуће. Стога је терапија у овом случају углавном успорити процес пада вида, а такође и компензовати губитак слуха. Могући третмани укључују:
- Употреба групе витамина А (неки офталмологи верују да високе дозе палмитата витамина А могу успорити, али не и зауставити, прогресију ретинитиса пигмента);
- Имплантација специјалних електронских уређаја у оријентима пацијента (слушни апарат, кохлеарни имплантати.
Офталмолози препоручују да већина одраслих пацијената са напредним обликом пигмента ретинитису узима 15.000 ИУ (међународних јединица) дневног витамина А у облику палмитата под надзором. Пошто људи са синдромом типа 1 Усхер нису учествовали у студији, високе дозе витамина А се не препоручују за ову групу пацијената. Људи који размишљају о узимању витамина А требају разговарати о овој опцији са својим доктором. Остале препоруке које се односе на ову опцију третмана укључују:
- Промените исхрану уз укључивање хране високе количине витамина А.
- Жене које планирају трудноћу треба престати узимати високе дозе витамина А три месеца прије планиране концепције због повећаног ризика од порођаја.
- Жене које су трудне престану узимати високе дозе витамина А због повећаног ризика од порођаја.
Такође је важно прилагодити такво дијете друштвеном животу. То захтева помоћ едукатора-дефектолога, као и психолога. У случају када пацијент започне прогресиван пад визије, треба га научити да користи знаковни језик.
Прогноза
Усхеров синдром има неповољну прогнозу. Видно поље и његова озбиљност почињу да се погоршавају у периоду од 20-30 година код већине пацијената са било којом врстом болести. У неким случајевима долази до потпуне билатералне губитке вида. Глухом, која се увек примећује и неумност, врло брзо се развија до пуне билатералне губитке слуха.