Медицински стручњак за чланак

Нове публикације
Аперов синдром
Последње прегледано: 04.07.2025

Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.

 [
[ Узроци Аперов синдром
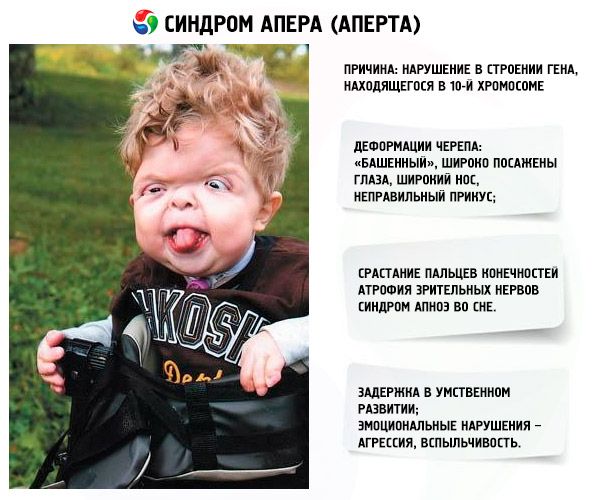
Развој синдрома изазива поремећај у структури гена који се налази на хромозому 10. Овај ген је одговоран за процес раздвајања прстију, а поред тога и за благовремено затварање кранијалних шавова.
Поред тога, заразне болести прележане током трудноће (као што су рубеола или туберкулоза, сифилис или грип, као и менингитис), као и излагање мајке рендгенским зрацима сматрају се узроцима патологије. Најчешће се такав синдром открива код деце рођене од старијих родитеља.
Патогенеза
Апертов синдром је наследан. Његов тип је аутозомно доминантан (тј. у породици у којој је један од будућих родитеља болестан, вероватноћа да ће беба имати ову болест је 50-100%).
Јединствена мутација у рецептору 2 фактора раста фибробласта (FGFR2) доводи до повећаног броја ћелија прогенитора које се развијају дуж остеогеног пута. Ово на крају резултира повећаним формирањем субпериосталног коштаног матрикса и превременом осификацијом кранијалних шавова током феталног развоја. Редослед и брзина топљења шавова одређују степен деформитета и инвалидитета. Када се материјал за шав зацели, раст других ткива управно на овај шав је ограничен, а срасле кости делују као јединствена коштана скела.
Први генетски доказ да је синдактилија код Апертовог синдрома последица дефекта рецептора фактора раста кератиноцита (KGFR) био је посматрање корелације између експресије KGFR у фибробластима и тежине синдактилије.
Амблиопија и страбизам су чешћи код пацијената са мутацијом FGFR2 Ser252Trp, а атрофија оптичког диска је чешћа код пацијената са мутацијом FGFR2 Pro253Arg. Пацијенти са мутацијама FGR2 Ser252Trp имају значајно већу преваленцију оштећења вида у поређењу са пацијентима са мутацијом FGFR2 Pro253Arg.
Симптоми Аперов синдром
Неке манифестације болести су јасно приметне већ при рођењу бебе, јер се развијају у мајчиној утроби. Међу главним симптомима синдрома су:
- Лобања је деформисана - протеже се у висину, добијајући изглед „куле“; поред тога, очи су широко постављене и благо испупчене (јер се величина очних дупљи смањује); нос се шири и формира се неправилан загриз (горњи зуби прекомерно штрче);
- Прсти удова су потпуно срасли (углавном домали прст, као и средњи прст и кажипрст) и изгледају као кожна мембрана или потпуно срастање костију; поред тога, могу израсти и додатни прсти;
- Ментална ретардација (не јавља се код свих);
- Оптички живци атрофирају, због чега се смањује оштрина вида (у неким случајевима се развија потпуни губитак вида);
- Повећан интракранијални притисак, који настаје као резултат превременог затварања кранијалних шавова, манифестује се у облику главобоље и повраћања са мучнином;
- Пошто горња вилица остаје неразвијена, настају проблеми са дисањем;
- Синдром апнеје у сну је честа појава.
- Емоционалне манифестације – агресија, недостатак контроле, јака раздражљивост.
Дијагностика Аперов синдром
Да би се поставила дијагноза, потребни су следећи кораци:
- Лекар треба да анализира жалбе пацијента, као и медицинску историју. Потребно је сазнати да ли је било случајева сличне патологије у породици;
- Неуролошки преглед за процену облика лобање и интелектуалног развоја пацијента (специјални упитници као и интервју);
- Преглед фундуса ради откривања присуства симптома повећаног интракранијалног притиска (отицање оптичког диска, као и замућење његових ивица);
- Да би се проценило стање лобање, врши се рендгенски снимак;
- Компјутерска и магнетна резонанца главе за испитивање структуре мозга и лобање слој по слој, утврђивање присуства симптома превременог срастања кранијалних шавова, а поред тога и хидроцефалуса (због повећаног интракранијалног притиска, акумулира се вишак цереброспиналне течности (то је цереброспинална течност која промовише метаболички процес, као и исхрану мозга));
- Рентген стопала и руку ради утврђивања узрока спајања прстију (ово је важно за планирање накнадне хируршке интервенције);
- Може се прописати консултација са медицинским генетичарем и неурохирургом.
Тестови
Спроводи се генетска анализа уобичајених мутација које се јављају у гену типа FGFR2.
Диференцијална дијагноза
Овај синдром се мора разликовати од других генетских патологија код којих се примећује краниосиностоза. То су болести као што су Фајферов, Крузонов, Саетре-Шоценов и Карпентеров синдром. Молекуларно-генетске методе тестирања се користе за искључивање ових аномалија.
Кога треба контактирати?
Третман Аперов синдром
Хирургија се сматра јединим ефикасним третманом за Апертов синдром - помаже у исправљању индивидуалних физичких недостатака, а такође исправља и менталну ретардацију.
Током ове процедуре, коронални шав се затвара како би се спречила могућа повреда мозга. Најчешћа техника је краниофацијална дистракција, која подразумева постепену тракцију лобање. Ортодонтска и/или ортогнатска хирургија се изводи ради уклањања појединачних дефеката лица.
Поред тога, пацијенти се подвргавају хируршкој корекцији фузије прстију.
Референце
Медицинска генетика. Национални водич. Уредио Гинтер ЕК, Пузирев ВП ГЕОТАР-Медиа. 2022