
Мартин-Беллов синдром
Последње прегледано: 23.04.2024

Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.
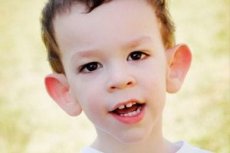
Мартин-Бела синдром описан је 1943. Године. Доктори, чије су имена именоване. Болест је генетски поремећај, који се састоји од менталне ретардације. 1969 откривене су промене карактеристичне за ову болест у хромозому Кс (крхкост у дисталном рамену). 1991. Године Научници су открили ген који је одговоран за развој ове болести. Ова болест се назива и "крхки хромозом-Кс синдром". Болест погађа и дечаке и девојчице, али чешће (3 пута) дечаци су болесни.
 [
[Епидемиологија
Мартин-Белла синдром је прилично честа болест: постоји 0,3-1,0 људи који болују од ове болести на 1000 мушкараца, и 0,2-0,6 на 1000 жена. А дјеца с Мартин-Белловим синдромом су рођена на свим континентима са истом фреквенцијом. Очигледно је да националност, боја коже, рез очију, услови живота, добробит људи не утичу на почетак болести. Учесталост њеног појаве може се упоредити само са учесталошћу Довновог синдрома (за 600-800 новорођенчади, 1 болест). Петина мушких носиоца промењеног гена су здраве, немају клиничке и генетске абнормалности, а остало са знацима менталне ретардације од благих до тешких облика. Међу женским носиоцима пацијената нешто више од трећине.
Крхки Кс хромосомски синдром утјече на отприлике 1 у 2500-4000 мушкараца и 1 у 7000-8000 жена. Преваленција носиоца болести међу женском популацијом процењује се на 1 на 130-250 људи; Проценат преовлађивања носилаца међу мушкарцима је 1 на 250-800.
Узроци мартин-Беллов синдром
Мартин-Белла синдром се развија услед потпуног или делимичног прекида производње специфичног протеина од стране тела. Ово је због недостатка одговора из гена попут ФМР1, локализованог у Кс хромосому. Мутација се јавља као резултат преуређивања структуре гена из нестабилних структурних варијанти генских стања (алела), а не од самог почетка. Болест се преноси само преко мушке линије, чиме човјек можда није неопходно болестан. Мале носиоци преносе ген у своје кћерке у непромењеном облику, тако да њихова ментална ретардација није очигледна. Са даљим преносом гена од мајке на своју дјецу, ген мутира, а затим се појављују сви знаци карактеристични за ову болест.
Патогенеза
Су мутације гена у апарату основу патогенези синдрома Мартин-Белл, што доводи до блокаде производње ФМР-протеина, протеина, животним телом, посебно у неурона и присутна у различитим ткивима. Студије показују да ФМР протеини су директно укључени у регулацију превода који се јављају у ткивима мозга. Одсуство овог протеина или његова ограничена продукција од стране тела доводи до менталне ретардације.
У патогенези болести, кључна повреда је хиперметилација гена, али није било могуће коначно одредити механизам развоја овог поремећаја.
Поред тога, откривена је локусна хетерогеност патологије, која је повезана са полиализмом, као и полилокусирањем. Утврђено је присуство алелних варијанти развоја болести, које су узроковане постојањем тачких мутација, као и уништавање ФМРЛ гена.
Такође код пацијената, детектују се 2 осетљива на фрагментарне триплете фолне киселине лоциране у З00 кб, а такође и 1,5-2 тп. Из крхког триплета, који садржи ген ФМР1. Механизам јавља у генима ФРАКСЕ и ФРАКСФ (они су идентификовани у горе крхки триплета) мутације корелацији са механизмом поремећаја синдром Мартин Белл. Овај механизам је услед пропагације ГЦЦ-а, као и ЦГГ-понављања, под којима се јавља метилација тзв. ЦпГ-острва. Поред класичног облика патологије, постоје и два ретка типа која се разликују због експанзије тринуклеотидних понављања (у мушким и женским мејозама).
Откривено је да у класичном облику синдрома пацијент нема посебан нуклеоцитоплаземски протеин типа ФМР1, који обавља функцију везивања различитих мРНА. Поред тога, овај протеин унапређује формирање комплекса који помаже у извођењу транслационих процеса унутар рибозома.
Симптоми мартин-Беллов синдром
Како препознати болест код деце? Који су први знаци? У првим месецима живота детета, симптом Мартин-Белл-а се не може препознати, осим што понекад постоји и смањење мишићног тона. После годину дана, клиника болести је очигледнија: дете почиње да крене касније и говори, понекад говори потпуно одсутни. Хиперактиван је, насумично махну рукама, у страху од гужве и буке, тврдоглави, оштри избијања беса, емоционална нестабилност, епилептичне пригоде, не иде у контакт са очима. Код пацијената са синдромом Мартин-Белла, болест такође производи изглед: уши које штрче и велике, чело теже, издужено лице, брадавица која штрчи, страбизам, широке четке и стопала. За њих су карактеристични ендокринални поремећаји: често тешка тежина, гојазност, мушкарци имају велике тестисе, рани пубертет.

Међу пацијентима с Мартином-Белловим синдромом, ниво интелигенције је веома различит: од мале менталне ретардације до тешких случајева. Ако нормална особа има ИК од 100 у просјеку и геније од 130, онда је 35-70 људи у ризику од болести.
Сви клинички симптоми патологије могу се карактеризирати тријом основних манифестација:
- олигофренија (ИК је 35-50);
- дисморпхопхобиа (испадајуће уши, као и прогнатизам);
- макроорхидизма, који се манифестује после појаве пубертета.
Приближно 80% пацијената такође открива пролапс бицуспид вентила.
Али пуни облик синдрома се манифестује само у 60% свих пацијената. У 10% је пронађена само ментална ретардација, док се код других болест развија са другачијом комбинацијом симптома.
Међу првим знацима болести манифестоване су већ ране године:
- болесно дете има значајну менталну ретардацију у поређењу са развојем других вршњака;
- поремећаји пажње и фокуса;
- јака тврдоглавост;
- деца прилично касно почињу да ходају и причају;
- постоје хиперактивност и поремећаји у развоју говора;
- веома јаке и неконтролисане борбе беса;
- може развити мутизам - ово је потпуно одсуство дететовог говора;
- клинац осећа социјалну анксиозност, може да паника због гласне буке или других јаких звукова;
- дете неконтролисано и хаотично махне рукама;
- постоји стидљивост, дете се плаши да остаје на местима велике гомиле људи;
- појављивање разних опсесија, нестабилно емоционално стање;
- беба може бити невољна да успоставља контакт са људима.
Код одраслих примјећују се сљедећи симптоми патологије:
- специфичан изглед: издужено лице са тешким челом, велика удубљена ушеса, снажно испупчена брада;
- равне ноге, отитис и страбизам;
- пубертет се јавља прилично рано;
- гојазност може да се развије;
- прилично често у синдрому Мартина Бела постоје недостаци у развоју срца;
- код мушкараца постоји повећање тестиса;
- зглобови зглобова постају веома мобилни;
- Тежина, а такође и раст нагло повећава.
Дијагностика мартин-Беллов синдром
Да бисте дијагностиковали синдром Мартина Белла, потребно је да контактирате квалификованог генетичара. Дијагноза се врши након извођења специфичних генетских тестова, који омогућавају утврђивање дефектног хромозома.
Анализе
У раној фази развоја болести се користи цитогенетичка метода, у којој пацијент узима комад ћелијског материјала, на који се затим додаје фолна киселина како би изазвала промене у хромозомима. После одређеног временског периода, откривен је регион хромозома, на коме се примећује опазно прорезивање - то је знак присуства крхког Кс хромозомског синдрома.
Али ова анализа није погодна за дијагнозу у касним стадијумима болести, јер се његова тачност смањује услед широке употребе мултивитамина који садрже фолну киселину.
Интегрисана дијагноза Мартин-Белловог синдрома је молекуларно-генетски преглед који се састоји у одређивању броја такозваних тринуклеотидних понављања у гену.
Инструментална дијагностика
Високо специфична метода инструменталне дијагнозе је ПЦР (полимеразна ланчана реакција), која омогућава проучавање структуре амино киселинских остатака садржаних у Кс хромозому и тиме утврђује присуство Мартин Белл синдрома.
Постоји и посебан, још специфичнији метод за дијагностиковање патологије - комбинација ПЦР-а и детекције помоћу капиларне електрофорезе. Ова метода је изузетно тачна и открива хромозомску патологију код пацијената са примарним обликом инсуфицијенције јајника, као и атаксичног синдрома.
Утврђивање присуства дефекта може бити након дијагнозе на ЕЕГ-у. Код пацијената са овом болести примећена је слична биоелектрична активност мозга.
Који су тестови потребни?
Диференцијална дијагноза
Диференциране методе које помажу у сумњи у синдром укључују:
- клинички - 97,5% пацијената има очигледне знаке менталне ретардације (умерено или дубоко); 62% је имало велике уши; 68,4% има истакнуто видно чело и чело; у 68,4% дечака - повећавају се тестиси, у 41,4% - особине говора (брзина говора је неуједначена, гласност се не може контролисати итд.);
- цитогенетички - крв се испитује за културу лимфоцита, одређује се број ћелија с прекидом Кс хромозома на 100 испитаних ћелија;
- електроенцефалографија - промене у електричним импулсима мозга су специфичне за Мартин-Беллов синдром.
Кога треба контактирати?
Третман мартин-Беллов синдром
Антидепресиви са психостимулансима се користе у лечењу одраслих пацијената. Процес терапије лековима стално надзире психолог и психијатар. Поред тога, приватне клинике обављају поступке микроинжења са лековима као што су Церебролисин (или његови деривати), као и цитомедини (као што је Солцосерил или Лидасе).
Са развојем атаксичног синдрома, користе се лекови који разблажују крв, као и ноотропије. Поред тога, прописују се смеше амино киселина и ангиопротектори. Женама са примарним обликом јачине јајника прописани су корективни третман са фитофармантима и естрогеним.
Такође, у третману се користе антагонисти рецептора глутамина.
Традиционално за лечење Мартин-Белловог синдрома је употреба лекова који утичу на симптоме болести, али не због његовог узрока. Ова терапија се састоји у постављању антидепресива, неуролептика, психостимуланата. Нису сви лекови назначени за употребу код деце, тако да је листа лекова прилично ограничена. Неуролептици који се могу користити након 3 године (најранији узраст њиховог именовања) укључују халоперидол у капи и таблете, хлорпромазин у раствору, перикаазин у капи. Дакле, доза узимања халоперидола за децу израчунава се зависно од телесне тежине. За одрасле, доза се администрира појединачно. Прима се унутра, почне са 0,5-5 мг 2-3 пута дневно, онда се доза постепено повећава на 10-15 мг. Када се побољша, идите у нижу дозу да бисте одржали постигнуто стање. Са психомоторном стимулацијом одредити 5-10 мг интрамускуларно или интравенозно, могуће је неколико понављања у 30-40 минута. Дневна доза не би требало да прелази 100 мг. Могућа нежељена дејства у виду мучнине, повраћања, грчева, повећаног притиска, аритмије итд. Посебне мјере опреза требају пратити старији људи. Пријављени су случајеви изненадног срчане акције и може се појавити касна дискенезија (појављивање нехотичних покрета).
Антидепресиви повећавају активност можданих структура, олакшавају депресивно расположење, напетост, повећавају расположење. Ови лекови, препоручени за примање од 5-8 година са Мартин-Белловим синдромом, укључују кломипромин, сертралин, флуксегин, флувоксамин. Дакле, флуоксетин се узима током оброка унутар 1-2 (пожељно ујутру), почиње са 20 мг дневно, повећава се на 80 мг ако је потребно. Старији људи не препоручују дозу већу од 60 мг. Ток лијечења одређује лекар, али не више од 5 недеља.
Могуће нежељене реакције: вртоглавица, анксиозност, тинитус, смањење апетита, тахикардија, оток итд. Потребно је водити рачуна о именовању старијих особа са кардиоваскуларним болестима, дијабетес мелитусом.
Психостимуланти - психотропни лекови, који се користе за побољшање перцепције спољашњих стимулуса: погоршавају слух, одговор, визију.
Као седатив са неурозама, анксиозност, епилептички напади, епилептици, диазепам је прописан. Узима се орално, интравенозно, интрамускуларно, ректално (у ректум). Препоручује се појединачно, у зависности од тежине болести, од најмањих доза од 5-10 мг, дневно - 5-20 мг. Трајање лечења је 2-3 месеца. За децу се доза израчунава узимајући у обзир телесну тежину и индивидуалне карактеристике. Нежељени ефекти укључују летаргију, апатију, поспаност, мучнину, запрту. Опасно је комбиновати са алкохолом, евентуално заснованом на леку.
Код лечења Мартин-Белловог синдрома случајеви побољшања стања су такође забележени када су направљене лекове засноване на животињском материјалу (мозгу): церебролисате, церебролисин, церебролизате-М. Главне компоненте ових лекова су пептиди, који доприносе стварању протеина у неуронима, чиме се допуњују недостајући протеини. Церебролисин убризгава се на 5-10 мл, током терапије се састоји од 20-30 ињекција. Деца преписују лек из доба живота, интрамускуларно убризгавају сваки дан за 1-2 мл током месеца. Могуће поновљене сесије пријема. Нежељени ефекти у облику топлоте, контраиндиковани код трудница.
Било је покушаја лечења болести са фолном киселином, али је побољшан само бихевиорални аспект (ниво агресије, смањена хиперактивност, побољшани говор), али на интелектуалном нивоу ништа се није променило. За побољшање стања болести прописана фолна киселина, назначене су методе физиотерапије, логопедске, педагошке и социјалне корекције.
Литијумски препарати се такође сматрају ефикасним, што помаже побољшању прилагођавања пацијента у друштвеном окружењу, као и когнитивној активности. Поред тога, и даље регулишу његово понашање у друштву.
Употреба биља у Мартин-Белловом синдрому је могућа као антидепресиви. Биљке које помажу у смањивању напетости, анксиозности, побољшању сна укључују валеријску, мопарску, тимијанску, шентјанжеву, камилицу. Инфузије се припремају на следећи начин: 1 кашичицу из сушених биљака требаће се чаша воде која се загреје, инфузије инсистирају најмање 20мин, узимају се претежно ноћу пре одласка у кревет или поподне. Добар додатак њима биће једна жлица меда.
Физиотерапеутски третман
Да би се елиминисале неуролошке манифестације, извршене су посебне физиотерапијске процедуре - као што су вјежбе у базенима, релаксација мишића и акупунктура.
Оперативни третман
Важну фазу лечења се такође сматрају методама пластичне хирургије - операцијама које помажу у побољшању изгледа пацијента. Пластика екстремитета и ушица, а поред ових гениталија се изводе. Постоји и корекција гинекомастије са еписпадијом, а уз то и друге недостатке изгледа.
Превенција
Једини начин превенције болести је пренатални преглед трудница. Постоје посебни тестови који омогућавају рано откривање присуства патологије, након чега се препоручује прекид трудноће. Као алтернатива, користи се ИВФ који може да помогне детету да наследи здрав хромозом Кс.
Профилакса пацијента зависи од тога да ли се мутација гена поново појавила или је наследила. За ово се спроводи молекуларна генетичка дијагноза. У корист "свежине" мутације, чињеница да рођаци нису открили "крхки Кс-хромозом", сведочи да је ризик да се дете са Мартин-Белловим синдромом веома мала. У породицама у којима постоје пацијенти, тест ће помоћи да се избегну поновљени случајеви.
Прогноза
Прогноза Мартин-Белловог синдрома за живот је повољна, за опоравак - не. Очекивано трајање живота зависи од тежине болести и пратећих порока. Пацијент може живети у уобичајеном трајању. У тешким облицима Мартин-Белловог синдрома, смртно опасна инвалидност угрожава пацијенте.
Животни вијек
Мартин Беллов синдром нема озбиљан негативан утицај на здравље, па је очекивани животни век већине људи који су пронашли ову патологију, не разликује од стандардних индикатора.