Медицински стручњак за чланак

Нове публикације
Мартин-Белов синдром
Последње прегледано: 04.07.2025

Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.
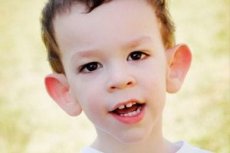
Мартин-Белов синдром су описали лекари 1943. године, по којима је и добио име. Болест је генетски поремећај који се састоји од менталне ретардације. Године 1969. идентификоване су промене на хромозому X (крхкост у дисталном делу крака) карактеристичне за ову болест. Године 1991. научници су открили ген одговоран за развој ове болести. Ова болест се назива и „синдром крхког X“. И дечаци и девојчице су подложни болести, али дечаци су чешће (3 пута) погођени.
Епидемиологија
Мартин-Белов синдром је прилично честа болест: 0,3-1,0 од 1.000 мушкараца пати од ове болести, а 0,2-0,6 од 1.000 жена. Штавише, деца са Мартин-Беловим синдромом рађају се на свим континентима са истом учесталошћу. Очигледно је да националност, боја коже, облик очију, услови живота и благостање људи не утичу на појаву болести. Њена учесталост појаве је упоредива само са учесталошћу Дауновог синдрома (1 болест од 600-800 новорођенчади). Петина мушких носилаца измењеног гена је здрава, нема клиничких или генетских абнормалности, остали имају знаке менталне ретардације од благих до тешких облика. Међу женским носиоцима, нешто више од трећине је болесно.
Синдром фрагилног X хромозома погађа приближно 1 од 2.500–4.000 мушкараца и 1 од 7.000–8.000 жена. Преваленција носилаштва међу женама процењује се на 1 од 130–250; преваленција носилаштва међу мушкарцима процењује се на 1 од 250–800.
Узроци Мартин-Белов синдром
Мартин-Белов синдром се развија због потпуног или делимичног престанка производње специфичног протеина у телу. Ово се дешава због недостатка одговора гена FMR1, локализованог у X хромозому. Мутација настаје као резултат реструктурирања гена из нестабилних структурних варијанти генских стања (алела), а не од самог почетка. Болест се преноси само по мушкој линији, а мушкарац не мора нужно бити болестан. Мушки носиоци преносе ген на своје ћерке у непромењеном облику, тако да њихова ментална ретардација није очигледна. Са даљим преношењем гена са мајке на њену децу, ген мутира, и појављују се сви знаци карактеристични за ову болест.
Патогенеза
Патогенеза Мартин-Беловог синдрома заснива се на мутацијама генског апарата, које доводе до блокирања производње FMR протеина, протеина виталног за тело, посебно у неуронима, а присутан је у различитим ткивима. Истраживања показују да су FMR протеини директно укључени у процесе регулације транслација који се дешавају у можданом ткиву. Одсуство овог протеина или његова ограничена производња од стране тела доводи до менталне ретардације.
У патогенези болести, хиперметилација гена се сматра кључним поремећајем, али још увек није било могуће дефинитивно идентификовати механизам развоја овог поремећаја.
Истовремено, откривена је и локусна хетерогеност патологије, која је повезана са полиалелизмом, као и полилокус. Утврђено је присуство алелних варијанти развоја болести, које су узроковане постојањем тачкастих мутација, као и уништењем гена типа FMRL.
Пацијенти такође имају 2 крхка триплета осетљива на фолну киселину, смештена 300 kb, као и 1,5-2 милиона bp из крхког триплета који садржи ген FMR1. Механизам мутација које се јављају у генима FRAXE и FRAXF (идентификовани су у горе поменутим крхким триплетима) повезан је са механизмом поремећаја код Мартин Беловог синдрома. Овај механизам је узрокован ширењем GCC и CGG понављања, која изазивају метилацију такозваних CpG острва. Поред класичног облика патологије, постоје и 2 ретка типа која се разликују због експанзије тринуклеотидних понављања (у мушкој и женској мејози).
Утврђено је да код класичног облика синдрома пацијенту недостаје посебан нуклеоцитоплазматски протеин типа FMR1, који обавља функцију везивања различитих мРНК. Поред тога, овај протеин промовише формирање комплекса који помаже у спровођењу процеса транслације унутар рибозома.
Симптоми Мартин-Белов синдром
Како препознати болест код деце? Који су први знаци? У првим месецима живота детета немогуће је препознати Мартин-Белов симптом, осим што се понекад примећује смањење мишићног тонуса. После годину дана, клиничка слика болести је очигледнија: дете почиње касно да хода и говори, понекад је говор потпуно одсутан. Хиперактивно је, насумично маше рукама, плаши се гужве и буке, тврдоглаво је, јављају се оштри изливи беса, емоционална нестабилност, јављају се епилептични напади, не успоставља контакт очима. Код пацијената са Мартин-Беловим синдромом, болест се одаје и по изгледу: уши су штрчеће и велике, чело је тешко, лице је издужено, брада је истурена, страбизам, широке руке и стопала. Карактеришу их и ендокрини поремећаји: често велика тежина, гојазност, велики тестиси код мушкараца, рани пубертет.

Међу пацијентима са Мартин-Беловим синдромом, ниво интелигенције значајно варира: од благе менталне ретардације до тешких случајева. Ако нормална особа има коефицијент интелигенције (IQ) у просеку 100, а геније 130, онда људи подложни болести имају 35-70.
Сви клинички симптоми патологије могу се окарактерисати тријадом главних манифестација:
- олигофренија (IQ је 35-50);
- дисморфофобија (примећују се штрчеће уши и прогнатизам);
- макроорхидизам, који се појављује након почетка пубертета.
Приближно 80% пацијената такође има пролапс бикуспидалног залистка.
Међутим, пуни облик синдрома се манифестује само код 60% свих пацијената. Код 10% се открива само ментална ретардација, а код осталих се болест развија са другачијом комбинацијом симптома.
Међу првим знацима болести, који се јављају у раном добу:
- болесно дете показује значајну менталну ретардацију у поређењу са развојем других вршњака;
- поремећаји пажње и концентрације;
- јака тврдоглавост;
- деца почињу да ходају и говоре прилично касно;
- примећују се хиперактивност и поремећаји развоја говора;
- веома јаки и неконтролисани напади беса;
- може се развити мутизам - ово је потпуни недостатак говора код детета;
- беба доживљава социјалну анксиозност и способна је да паничи због гласне буке или било којих других гласних звукова;
- дете неконтролисано и хаотично маше рукама;
- примећује се стидљивост, дете се плаши да буде у гужви;
- појава разних опсесивних идеја, нестабилно емоционално стање;
- Беба може бити невољна да успостави контакт очима са људима.
Код одраслих се примећују следећи симптоми патологије:
- специфичан изглед: издужено лице са тешким челом, велике штрчеће уши, снажно штрчећа брада;
- равна стопала, отитис и страбизам;
- пубертет се јавља прилично рано;
- може се развити гојазност;
- Често се срчани дефекти примећују код Мартин-Беловог синдрома;
- код мушкараца се примећује повећање тестиса;
- артикулације зглобова постају веома покретне;
- тежина и висина нагло повећавају.
Дијагностика Мартин-Белов синдром
Да бисте дијагностиковали синдром Мартина Бела, потребно је да контактирате квалификованог генетичара. Дијагноза се поставља након специфичних генетских тестова који вам омогућавају да идентификујете дефектни хромозом.
Тестови
У раној фази болести користи се цитогенетска метода, којом се од пацијента узима фрагмент ћелијског материјала, коме се затим додаје фолна киселина како би се изазвале промене у хромозомима. Након одређеног временског периода, идентификује се подручје хромозома где је приметно проређивање - то је знак присуства синдрома фрагилног X хромозома.
Међутим, овај тест није погодан за дијагнозу у каснијим фазама болести, јер је његова тачност смањена широком употребом мултивитамина који садрже фолну киселину.
Интегрисана дијагностика Мартин-Бел синдрома је молекуларно-генетски преглед, који се састоји у одређивању броја такозваних тринуклеотидних понављања у гену.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Инструментална дијагностика
Веома специфична метода инструменталне дијагностике је ПЦР (полимеразни ланчани реактор), која омогућава проучавање структуре аминокиселинских остатака садржаних у Кс хромозому и тиме утврђивање присуства Мартин Беловог синдрома.
Постоји и посебна, још специфичнија, метода дијагностике патологије – комбинација ПЦР-а и детекције помоћу капиларне електрофорезе. Ова метода је веома прецизна и открива хромозомску патологију код пацијената са примарном инсуфицијенцијом јајника, као и атаксичним синдромом.
Присуство дефекта може се утврдити након обављања дијагностике на ЕЕГ-у. Пацијенти са овом болешћу имају сличну биоелектричну активност мозга.
Који су тестови потребни?
Диференцијална дијагноза
Диференциране методе које помажу у сумњи на синдром укључују:
- клинички - 97,5% пацијената има очигледне знаке менталне ретардације (умерене или дубоке); 62% има штрчеће велике уши; 68,4% има велику штрчећу браду и чело; 68,4% дечака има увећане тестисе, 41,4% има говорне особености (неуједначен темпо говора, неконтролисана јачина звука итд.);
- цитогени - крв се испитује на културу лимфоцита, одређује се број ћелија са крхким Кс хромозомом на 100 проучаваних ћелија;
- Електроенцефалографија - бележе се промене електричних импулса мозга које су специфичне за Мартин-Белов синдром.
Кога треба контактирати?
Третман Мартин-Белов синдром
У лечењу одраслих пацијената користе се антидепресиви са психостимулансима. Процес терапије лековима стално прати психолог и психијатар. Поред тога, у приватним клиникама се спроводе поступци микроињекције са лековима као што су Церебролизин (или његови деривати), као и цитомедини (као што су Солкосерил или Лидаза).
У развоју атаксичног синдрома користе се лекови за разређивање крви и ноотропици. Поред тога, прописују се мешавине аминокиселина и ангиопротектори. Женама са примарном инсуфицијенцијом јајника прописује се корективни третман употребом биљних лекова и естрогена.
Антагонисти глутаминских рецептора се такође користе у лечењу.
Традиционално, лечење Мартин-Беловог синдрома подразумева употребу лекова који утичу на симптоме болести, али не и на њен узрок. Ова терапија подразумева прописивање антидепресива, неуролептика и психостимуланата. Нису сви лекови индиковани за употребу код деце, тако да је листа лекова прилично ограничена. Неуролептици који се могу користити након 3 године (најранији узраст за њихово прописивање) укључују халоперидол у капима и таблетама, хлорпромазин у раствору и перициазин у капима. Дакле, доза халоперидола за децу се израчунава у зависности од телесне тежине. За одрасле, доза се прописује појединачно. Узима се орално, почевши од 0,5–5 мг 2–3 пута дневно, затим се доза постепено повећава на 10–15 мг. Када дође до побољшања, прелазе на нижу дозу да би се одржало постигнуто стање. У случају психомоторне агитације, прописује се 5-10 мг интрамускуларно или интравенозно, могуће је неколико понављања након 30-40 минута. Дневна доза не сме прећи 100 мг. Могући су нежељени ефекти у облику мучнине, повраћања, грчева мишића, повећаног притиска, аритмије итд. Старије особе треба да предузму посебне мере предострожности, јер су регистровани случајеви изненадног срчаног застоја, а може доћи и до тардивне дискинезије (невољних покрета).
Антидепресиви повећавају активност можданих структура, ублажавају депресију, напетост и побољшавају расположење. Ови лекови, препоручени за употребу од 5-8 година код Мартин-Бел синдрома, укључују кломипромин, сертралин, флуоксетин и флувоксамин. Дакле, флуоксетин се узима орално током оброка 1-2 пута (пожељно у првој половини дана), почевши од 20 мг дневно, повећавајући на 80 мг ако је потребно. Старијим особама се не препоручује доза већа од 60 мг. Ток лечења одређује лекар, али не дуже од 5 недеља.
Могући нежељени ефекти: вртоглавица, анксиозност, тинитус, губитак апетита, тахикардија, едем итд. Потребан је опрез приликом прописивања старијим особама, особама са кардиоваскуларним болестима и дијабетесом.
Психостимуланси су психотропни лекови који се користе за побољшање перцепције спољашњих стимулуса: изоштравају слух, реакције и вид.
Диазепам се прописује као седатив за неурозе, анксиозност, епилептичке нападе и конвулзије. Узима се орално, интравенозно, интрамускуларно, ректално (у ректум). Прописује се појединачно, у зависности од тежине болести, са најмањим дозама од 5-10 мг, дневно - 5-20 мг. Трајање лечења је 2-3 месеца. За децу, доза се израчунава узимајући у обзир телесну тежину и индивидуалне карактеристике. Нежељени ефекти укључују летаргију, апатију, поспаност, мучнину, затвор. Опасно је комбиновати са алкохолом, могућа је зависност од лека.
У лечењу Мартин-Бел синдрома, забележени су случајеви побољшања стања и уз увођење лекова направљених од животињског материјала (мозга): церебролизат, церебролизин, церебролизат-М. Главне компоненте ових лекова су пептиди који подстичу производњу протеина у неуронима, чиме надокнађују недостајући протеин. Церебролизин се примењује млазом од 5-10 мл, ток лечења се састоји од 20-30 ињекција. Лек се прописује деци од годину дана, примењује се интрамускуларно сваког дана 1-2 мл током месец дана. Могуће су поновљене сесије примене. Нежељени ефекти у облику грознице, контраиндиковани за труднице.
Било је покушаја лечења болести фолном киселином, али се побољшао само аспект понашања (смањен је ниво агресије и хиперактивности, побољшан говор), а ништа се није променило на интелектуалном нивоу. За побољшање стања болести прописује се фолна киселина, индикују се методе физиотерапије, логопедска терапија, педагошка и социјална корекција.
Литијумски препарати се такође сматрају ефикасним, јер помажу у побољшању адаптације пацијента на друштвено окружење, као и когнитивне активности. Поред тога, регулишу и његово понашање у друштву.
Употреба биљака за Мартин-Белов синдром је могућа као антидепресиви. Биљке које помажу у ублажавању напетости, анксиозности и побољшању сна укључују валеријану, нану, тимијан, кантарион и камилицу. Инфузије се припремају на следећи начин: за 1 кашичицу сувог биља потребна је чаша кључале воде, декокције се инфузирају најмање 20 минута, узимају се углавном увече пре спавања или поподне. Кашика меда би била добар додатак њима.
Физиотерапијски третман
Да би се елиминисале неуролошке манифестације, спроводе се посебне физиотерапеутске процедуре - као што су вежбе у базену, опуштање мишића и акупунктура.
Хируршко лечење
Важном фазом лечења сматрају се и методе пластичне хирургије - операције које помажу у побољшању изгледа пацијента. Изводи се пластична хирургија удова и ушних шкољки, као и гениталија. Такође се врши корекција гинекомастије са еписпадијом, као и других недостатака у изгледу.
Превенција
Једини метод превенције болести је пренатални скрининг трудница. Постоје посебни прегледи који омогућавају рано откривање патологије, након чега се препоручује прекид трудноће. Као алтернатива, користи се вантелесна оплодња (IVF), која може помоћи детету да наследи здрав X хромозом.
Превенција пацијента зависи од тога да ли се генска мутација поново појавила или је наследна. За то се спроводи молекуларно-генетска дијагностика. Чињеница да тест није открио „фрагилни X хромозом“ код рођака говори у прилог „свежине“ мутације, што значи да је ризик од рађања детета са Мартин-Беловим синдромом веома мали. У породицама где има болесних људи, тест ће помоћи да се избегну поновљени случајеви.
Прогноза
Прогноза за Мартин-Белов синдром је повољна за живот, али не и за опоравак. Очекивани животни век зависи од тежине болести и пратећих дефеката. Пацијент може живети нормалним животом. Код тешких облика Мартин-Беловог синдрома, пацијенти су у ризику од доживотне инвалидности.
Очекивани животни век
Мартин Белов синдром нема озбиљан негативан утицај на здравље, тако да се животни век већине људи којима је дијагностикована ова патологија не разликује од стандардних индикатора.