Медицински стручњак за чланак

Нове публикације
Синдром Тричера Колинса
Последње прегледано: 04.07.2025

Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.

Интраутерини поремећаји у процесима развоја костију узрокују озбиљне краниофацијалне деформације, а једна од варијанти такве патологије је синдром Тричера Колинса (ТКС) или мандибулофасцијална, односно максилофацијална дизостоза.
Шифра болести према МКБ 10: класа XVII (урођене аномалије, деформације и хромозомски поремећаји), Q75.4 - мандибулофацијална дизостоза.
Узроци Синдром Тричера Колинса
Овај синдром је добио име по изузетном британском офталмологу Едварду Тричеру Колинсу, који је описао главне карактеристике патологије пре више од сто година. Међутим, европски лекари ову врсту аномалије костију лица и вилице чешће називају Франческотијевом болешћу или синдромом - на основу опсежног истраживања швајцарског офталмолога Адолфа Франческотија, који је средином прошлог века увео термин „мандибулофасцијална дизостоза“. У медицинским круговима се користи и назив Франческоти-Колинсов синдром.
Синдром Тричера Колинса узрокован је мутацијама у гену TCOF1 (на локусу хромозома 5q31.3-33.3), који кодира нуклеоларни фосфопротеин одговоран за формирање краниофацијалног дела људског ембриона. Као резултат превременог смањења количине овог протеина, биогенеза и функције рРНК су поремећене. Према генетичарима из програма истраживања људског генома, ови процеси доводе до смањења пролиферације ембрионалних ћелија неуралног гребена - гребена дуж неуралног жлеба, који се током ембрионалног развоја затвара у неуралну цев.
Формирање ткива лица настаје услед трансформације и диференцијације ћелија горњег (главног) дела нервног гребена, које мигрирају дуж нервне цеви до подручја првог и другог бранхијалног лука ембриона. А недостатак ових ћелија узрокује краниофацијалне деформације. Критични период за појаву аномалија је од 18 до 28 дана након оплодње. По завршетку миграције ћелија нервног гребена (у четвртој недељи гестације), формирају се готово сва растресита мезенхимална ткива у пределу лица, која се касније (од 5 до 8 недеља) диференцирају у скелетна и везивна ткива свих делова лица, врата, гркљана, уха (укључујући и унутрашње ухо) и будућих зуба.
Патогенеза
Патогенеза Тричер Колинсовог синдрома је често фамилијарна, а аномалија се наслеђује аутозомно доминантно, мада постоје случајеви аутозомно рецесивног преноса дефекта (са мутацијама у другим генима, посебно, POLR1C и POLR1D). Најнепредвидљивија ствар код максилофацијалне дизостозе је то што деца наслеђују мутацију само у 40-48% случајева. То јест, код 52-60% пацијената узроци Тричер Колинсовог синдрома нису повезани са присуством аномалије у породици, већ се сматра да се патологија јавља као резултат спорадичних генских мутација de novo. Највероватније су нове мутације последице тератогених ефеката на фетус током трудноће.
Међу тератогеним узроцима овог синдрома, стручњаци наводе велике дозе етанола (етил алкохола), зрачење, дим цигарета, цитомегавирус и токсоплазму, као и хербициде на бази глифосата (Roundal, Glyfor, Tornado, итд.). А листа јатрогених фактора укључује лекове за акне и себореју са 13-цис-ретиноичном киселином (Isotretinoin, Accutane); антиконвулзивни лек Фенитоин (Dilantin, Epanutin); психотропне лекове Диазепам, Валијум, Реланијум, Седуксен.
Симптоми Синдром Тричера Колинса
Углавном, клинички знаци мандибулофасцијалне дизостозе и степен њихове експресије зависе од карактеристика манифестације генских мутација. А први знаци ове аномалије у већини случајева су видљиви код детета одмах након рођења: лице са Тричер Колинсовим синдромом има карактеристичан изглед. Штавише, морфолошке аномалије су обично билатералне и симетричне.
Најочигледнији симптоми синдрома Тричер Колинса су:
- неразвијеност (хипоплазија) костију лица лобање: зигоматични, зигоматични процеси фронталне кости, латералне птеригоидне плоче, параназални синуси, доња вилица и избочине коштаних епифиза (кондила);
- неразвијеност костију доње вилице (микрогнатија) и мандибуларни угао који је тупији него обично;
- нос је нормалне величине, али делује велико због хипоплазије суперцилијарних лукова и неразвијености или одсуства зигоматских лукова у темпоралном региону;
- очни прорези су усмерени надоле, односно облик очију је ненормалан, са спољним угловима који се спуштају надоле;
- дефекти доњих капака (колобом) и делимично одсуство трепавица на њима;
- неправилно обликоване ушне шкољке са широким спектром одступања, укључујући њихову локацију у углу доње вилице, одсуство режњева, слепе фистуле између трагуса уха и угла уста итд.;
- сужавање или затварање (атрезија) спољашњег слушног канала и аномалије средњег ува;
- одсуство или хипоплазија паротидних пљувачних жлезда;
- хипоплазија фарингеала (сужавање фарингеала и дисајних путева);
- несрастање тврдог непца (расцеп непца), као и одсуство, скраћивање или непокретност меког непца.
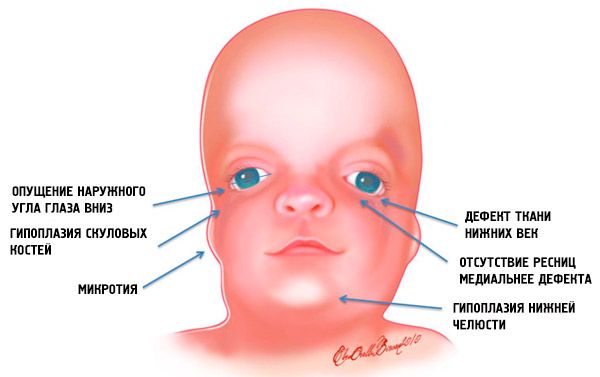
Такве анатомске аномалије у свим случајевима имају компликације. То су функционални поремећаји слуха у облику проводног губитка слуха или потпуне глувоће; оштећење вида услед неправилног формирања очних јабучица; дефекти непца узрокују тешкоће са храњењем и гутањем. Постоје поремећаји зубне оклузије (малоклузија) повезани са дефектима вилице, што, заузврат, узрокује проблеме са жвакањем и артикулацијом. Патологије меког непца објашњавају назални глас.
Компликације и посљедице
Последице максилофацијалних аномалија код Тричер Колинсовог синдрома су да су при рођењу интелектуалне способности детета нормалне, али због оштећења слуха и других поремећаја примећује се секундарна ментална ретардација.
Поред тога, деца са таквим манама акутно осећају своју инфериорност и пате, што негативно утиче на њихов нервни систем и психу.
Дијагностика Синдром Тричера Колинса
Постнатална дијагноза синдрома Тричер Колинса у суштини се заснива на клиничким знацима. Краниофацијална дизостоза се лако идентификује када је синдром потпуно изражен, али када су присутни минимално изражени симптоми патологије, могу се јавити проблеми са постављањем тачне дијагнозе.
У овом случају, посебну пажњу треба посветити процени свих функција повезаних са аномалијама, посебно оних које утичу на дисање (због ризика од апнеје у сну). Такође треба проценити и пратити ефикасност храњења и засићење хемоглобина кисеоником.
Касније, 5-6 дана након рођења, биће потребно утврдити обим оштећења слуха аудиолошким тестирањем, које треба обавити у породилишту.
Прописан је преглед, током којег се врши инструментална дијагностика флуороскопијом краниофацијалне дисморфологије; пантомографија (панорамски рендгенски снимак коштаних структура лобање лица); потпуна кранијална компјутеризована томографија у различитим пројекцијама; ЦТ или МРИ мозга за одређивање стања унутрашњег слушног канала.
Најранија – пренатална – дијагноза максилофацијалних аномалија у присуству Тричер Колинсовог синдрома у породичној историји могућа је биопсијом хорионских ресица у 10-11 недеља трудноће (поступак прети побачајем и инфекцијом материце).
Крвне анализе се такође узимају од чланова породице; у 16-17 недеља трудноће анализира се амнионска течност (трансабдоминална амниоцентеза); у 18-20 недеља трудноће се врши фетоскопија и узима се крв из феталних крвних судова плаценте.
Али најчешће се ултразвук користи у пренаталној дијагнози овог синдрома код фетуса (у 20-24 недељи трудноће).
Који су тестови потребни?
Диференцијална дијагноза
Исте методе користе специјалисти када је потребна диференцијална дијагностика да би се препознао благи синдром Тричер Колинса и разликовао од других конгениталних аномалија краниофацијалних костију, посебно: Апертовог, Крузоновог, Нагеровог, Петерс-Хевелсовог, Хелерман-Стефовог синдрома, као и хемифацијалне микросомије (Голденхаров синдром), хипертелоризма, превременог срастања кранијалних шавова (краниосиностоза) или оштећеног срастања костију лица (краниосиностоза).
Третман Синдром Тричера Колинса
Као и у свим случајевима генетски одређених конгениталних дефеката, лечење тешких облика Тричер Колинсовог синдрома је искључиво палијативно, јер једноставно не постоје терапијске методе за такве патологије. Спектар и степен деформација код овог синдрома су опсежни и, стога, природа и интензитет медицинске интервенције такође имају много опција.
Слушни апарати се користе за корекцију и побољшање слуха, а сеансе логопедске терапије за побољшање говора.
Хируршке интервенције су потребне у раном узрасту код тешких случајева сужавања дисајних путева (врши се трахеостомија) и ларинкса (врши се гастростомија ради храњења). Може бити потребна и хируршка корекција непца.
Операције продужења мандибуле се изводе у узрасту од 2-3 године или касније. Реконструкција меких ткива обухвата корекцију колобома доњег капка и аурикуларну пластичну хирургију.
Прогноза
Каква је прогноза за ову патологију? Зависи од степена деформације и интензитета симптома. Синдром Тричера Колинса је доживотна дијагноза.
 [ 25 ]
[ 25 ]