
Цхарцот-Марие-Тоотх болест
Последње прегледано: 23.11.2021

Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.
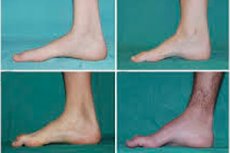
Атрофија, синдром перонеалног мишића или Цхарцот-Марие-Тоотх болест је читава група хроничних наследних болести са оштећењем периферних нерава.
Према ИЦД-10 у одељку болести нервног система, шифра ове болести је Г60.0 (наследна моторна и сензорна неуропатија). Такође је укључен у списак сироче болести.
Епидемиологија
Према клиничким статистикама, преваленција свих врста болести Цхарцот-Марие-Тоотх на 100 хиљада становника износи 19 случајева (према другим изворима, један случај на 2,5-10 хиљада становника).
ЦМТ тип 1 чини око две трећине случајева (један случај на 5-7 хиљада становника), а готово 70% њих повезано је са дуплирањем гена ПМП22. У свету ова врста болести погађа више од 1,2 милиона људи.
Инциденца ЦМТ типа 4 процењује се на 1-5 случајева на 10 хиљада деце. [1]
Узроци цхарцот-Марие-Тоотх болест
Према класификацији полинеуропатских синдрома , перонеална (перонеална) атрофија мишића, неурална амиотрофија Цхарцот-Марие-Тоотх-а или Цхарцот-Марие-Тоотх-ова болест (скраћено ЦМТ) односи се на генетски одређене моторно-сензорне полинеуропатије. [2]
Односно, разлози за његову појаву су генетске мутације. А у зависности од природе генетских абнормалности, разликују се главни типови или типови овог синдрома: демијелинизацијски и аксонски. У прву групу спада Цхарцот-Марие-Тоотх-ова болест (ЦМТ1), која се јавља као резултат дуплирања гена ПМП22 на хромозому 17, који кодира трансмембрански периферни протеин миелина 22. Као резултат, сегментна демијелинизација аксонске овојнице (процеси нервних ћелија) и долази до смањења брзине проводљивости нерва.сигнали. Поред тога, могу постојати мутације неких других гена.
Аксонски облик је Цхарцот-Марие-Тоотх болест тип 2 (ЦМТ2), који погађа саме аксоне и повезан је са патолошким променама у гену МФН2 у локусу 1п36.22, који кодира мембрански протеин митофусин-2, што је неопходно за фузију митохондрија и формирање функционалних митохондријских мрежа унутар периферних нерава ћелија. Постоји више од десетак подтипова ЦМТ2 (са мутацијама у одређеним генима).
Треба напоменути да је сада идентификовано више од стотину гена, чија штета, наследјена, узрокује различите подтипове болести Цхарцот-Марие-Тоотх. На пример, мутације у гену РАБ7 развијају тип 2Б ЦМТ; промена гена СХ3ТЦ2 (који кодира један од протеина шванских мембрана ћелије) узрокује ЦМТ типа 4Ц, који се манифестује у детињству и карактерише демијелинизација моторних и сензорних неурона (један и по туцети облика типа 4 ова болест се разликује).
Ретки СМТ типа 3 (назван Дејерине-Сотт синдром), изазван мутацијама гена ПМП22, МПЗ, ЕГР2 и других, такође почиње да се развија у раном детињству.
Када се ЦМТ тип 5 догоди у доби од 5 до 12 година, не примећује се само моторна неуропатија (у облику спастичне парапарезе доњих екстремитета), већ и оштећење оптичких и слушних живаца.
Слабост мишића и атрофија видног нерва (са губитком вида), као и проблеми са равнотежом, карактеристични су за ЦМТ тип 6. А код болести типа 7 Цхарцот-Марие-Тоотх, примећује се не само моторно-сензорна неуропатија, већ и мрежњача у облику пигментозног ретинитиса.
Чешћа СМТ везана за Кс или Цхарцот-Марие-Тоотх-ова болест са тетрапарезом екстремитета (слабљење кретања руку и ногу) међу мушкарцима је демијелинизирајућег типа и сматра се резултатом мутације гена ГЈБ1 на дугачак крак Кс хромозома, који кодира за Цоннекин 32, трансмембрански протеин Сцхванн ћелије и олигодендроците, који регулише пренос нервних сигнала. [3]
Фактори ризика
Главни фактор ризика за ЦМТ је присуство ове болести у породичној историји, односно код блиских рођака.
Према генетичарима, ако су оба родитеља носиоци аутосомно рецесивног гена Цхарцот-Марие-Тоотх болести, ризик од рођења детета код којег ће се развити ова болест износи 25%. А ризик да ће дете носити овај ген (али и сам неће имати никакве симптоме) процењује се на 50%.
У случају наслеђивања везаног за Кс (када је мутирани ген на женском Кс хромозому), постоји 50% ризика да мајка пренесе овај ген на свог сина, а он ће развити ЦМТ болест. Када се роди женско дете, болест се можда неће догодити, али синови ћерке (унуци) могу наследити оштећени ген - са развојем болести.
Патогенеза
Код било које врсте Цхарцот-Марие-Тоотх болести, његова патогенеза је последица наследне аномалије периферних живаца: моторних (моторних) и сензорних (сензорних).
Ако је ЦМТ тип демијелинизиран, тада уништавање или дефект мијелинске овојнице, која штити аксоне периферних нерава, доводи до успоравања преноса нервних импулса периферног нервног система - између мозга, мишића и сензорних органа.
У аксонском типу болести, аксони су директно погођени, што негативно утиче на снагу нервних сигнала, што је недовољно за потпуну стимулацију мишића и сензорних органа.
Такође прочитајте:
Како се шири Цхарцот-Марие-Тоотх синдром? Дефектни гени могу се наследити на аутосомно доминантан или аутосомно рецесиван начин.
Најчешће - аутосомно доминантно наследство - јавља се када постоји једна копија мутираног гена (коју носи један од родитеља). А вероватноћа преноса ЦМТ на свако рођено дете процењује се на 50%. [4]
У аутосомно рецесивном наследству, болест захтева две копије оштећеног гена (по једну од сваког родитеља који нема знакове болести).
У 40-50% случајева долази до аутосомно доминантне наследне демијелинизације, односно ЦМТ типа 1; у 12-26% случајева - аксонски ЦМТ, односно тип 2. И у 10-15% случајева се примећује наслеђивање везано за Кс. [5]
Симптоми цхарцот-Марие-Тоотх болест
Обично се први знаци ове болести почињу појављивати у детињству и адолесцентима и постепено се развијају током живота, мада се синдром може осетити и касније. Комбинација симптома је променљива, а стопа прогресије болести, као ни њена тежина, не могу се предвидети.
Постоје такви типични симптоми почетне фазе као повећани општи умор; смањени тонус (слабост) мишића стопала, зглобова и потколеница; недостатак рефлекса. То отежава кретање стопала и доводи до дисбазије (поремећаја хода) у виду веће коте ногу, често са честим спотицањем и падом. Знаци Цхарцот-Марие-Тоотх-ове болести код малог детета могу бити изражена неспретност и отежано ходање, неуобичајено за године старости, повезано са обостраним висећим стопалом . Карактеристични су и деформитети стопала: високи лук (шупље стопало) или јака равна стопала, закривљени (попут чекића) прсти.
У случају ходања на прстима на позадини мишићне хипотензије, неуролог може сумњати да дете има ЦМТ тип 4, у којем деца до адолесценције можда неће моћи да ходају.
Како напредују, атрофија и слабост мишића се шире на горње екстремитете, отежавајући фину моторику и нормалне активности руку. Смањење тактилних сензација и способност да се осећа топло и хладно, као и утрнулост стопала и руку, указује на оштећење аксона сензорних живаца.
Са манифестованом у детињству Цхарцот-Марие-Тоотх болешћу типа 3 и 6, постоји осетљива атаксија (поремећена координација покрета и равнотеже), трзање мишића и тремор, оштећење фацијалног нерва, атрофија оптичког црева са нистагмусом, губитак слуха.
У каснијим фазама може доћи до неконтролисаног дрхтања (подрхтавања) и честих грчева у мишићима; проблеми са кретањем могу довести до развоја болова: мишићних, зглобних, неуропатских.
Компликације и посљедице
Цхарцот-Марие-Тоотх болест може имати компликације и последице као што су:
- чешће угануће и преломи;
- контрактуре повезане са скраћивањем периартикуларних мишића и тетива;
- сколиоза (закривљеност кичме);
- проблеми са дисањем - са оштећењем нервних влакана која инервирају мишиће дијафрагме:
- губитак способности самосталног кретања.
Дијагностика цхарцот-Марие-Тоотх болест
Дијагностика укључује клинички преглед, историју (укључујући породичну историју), неуролошки и системски преглед.
Тестови се изводе ради провере опсега покрета, осетљивости и тетивних рефлекса. Провођење нерва се може проценити помоћу инструменталне дијагностике - електромиографије или електронеуромиографије . Такође ће бити потребан ултразвук или МРИ. [6]
Генетска или ДНК дијагностика за идентификовање најчешћих генетских мутација које узрокују ЦМТ на узорку крви су ограничене, јер ДНК тестови тренутно нису доступни за све врсте ЦМТ-а. За детаље погледајте - Генетичка истраживања
У неким случајевима се врши биопсија периферног нерва (обично гастроцнемиус).
Диференцијална дијагноза
Диференцијална дијагноза се врши са осталим периферним неуропатијама, Дуцхеннеовом мишићном дистрофијом, мијелопатичним и миастеничним синдромима, дијабетичном неуропатијом, мијелоаптијама у случају мултипле и амиотрофичне латералне склерозе, Гуиллаин-Барре синдрома, траумом перонеалног нерва и атрофијом диска кичме ), оштећење малог мозга или таламуса, као и нежељени ефекти хемотерапије (када се лече цитостатицима као што су винкристин или паклитаксел). [7]
Кога треба контактирати?
Третман цхарцот-Марие-Тоотх болест
Данас се лечење ове наследне болести састоји у физиотерапијским вежбама (усмереним на јачање и истезање мишића); радна терапија (која помаже пацијентима са слабостима мишића у рукама); користећи ортопедске уређаје за олакшавање ходања. Ако је потребно, узмите лекове против болова или антиконвулзиве. [8]
У случајевима изражених равних стопала може се извршити остеотомија, а у случају деформације пета индикована је њихова хируршка корекција - артродеза. [9]
У току су истраживања како генетске компоненте болести, тако и метода лечења. Употреба матичних ћелија, неких хормона, лецитина или аскорбинске киселине још увек није дала позитивне резултате.
Али захваљујући најновијим истраживањима, у блиској будућности, ново се заиста може појавити у лечењу болести Цхарцот-Марие-Тоотх. Дакле, од 2014. Године француска компанија Пхарнект развија, а од средине 2019. Године клиничка испитивања лека ПКСТ3003 за лечење ЦМТ типа 1 код одраслих, сузбијајући повећану експресију гена ПМП22, побољшавајући мијелинизацију периферних нерава и слабљење неуромускуларних симптома.
Специјалисти медицинске компаније Сарепта Тхерапеутицс (САД) раде на генској терапији за болест Цхарцот-Марие-Тоотх типа 1. Ова терапија ће користити безопасни адено-повезани вирус (ААВ) рода Депендовирус са линеарним једноланчаним ДНК геномом, који ће у тело пренети ген НТФ3, који кодира протеин неуротропин-3 (НТ-3) неопходан за функционисање Сцхванн-ових нервних ћелија.
До краја 2020. Године, Хеликмитх започиње клиничка испитивања генске терапије Енгенсис (ВМ202) развијене у Јужној Кореји за лечење мишићних симптома код типа 1 ЦМТ. [10]
Превенција
Превенција ЦМТ-а може бити генетско саветовање будућих родитеља, посебно ако неко из брачног пара има ову болест у породици. Међутим, идентификовани су случајеви мутација де ново генске тачке, односно у одсуству болести у породичној историји.
Током трудноће, узимање узорака хорионских ресица (од 10 до 13 недеља гестације), као и анализа плодне воде (у 15-18 недеља), омогућава вам да проверите вероватноћу појаве Цхарцот-Марие-Тоотх болести код нерођеног детета.
Прогноза
Генерално, прогноза за различите врсте Цхарцот-Марие-Тоотх болести зависи од клиничке тежине, али у сваком случају болест напредује полако. Многи пацијенти имају инвалидитет, иако то не смањује очекивани животни век.